Partager:



Syndrome de Rokitnasky-Küster-Hauser Symptômes, causes et traitements
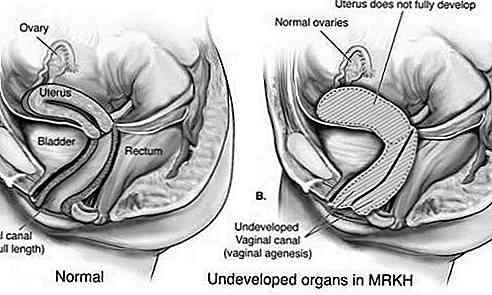
Syndrome de Rokitansky-Küster-Hauser (MRKH) est un trouble qui affecte le système reproducteur féminin caractérisé par un sous-développement ou une absence de l'utérus et du vagin.
Les femmes atteintes de ce syndrome développent des caractéristiques sexuelles secondaires pendant la puberté, les poils pubiens, mais n'ont pas de cycle menstruel (aménorrhée primaire).
Ne pas commencer le cycle menstruel est souvent le signe initial du syndrome MRKH. Bien que les femmes atteintes de cette maladie ne puissent pas avoir de grossesse, elles peuvent avoir des enfants par procréation assistée.
La gravité du syndrome peut varier selon le type. Le type 1 se caractérise par une absence isolée des deux tiers proximaux du vagin. Type II est caractérisé par d'autres défauts tels que des anomalies rénales (40% des cas), des anomalies squelettiques (20-25%), les troubles de l'audition (10%), et plus rarement, des anomalies cardiaques.
En raison de la nature de la maladie, elle est susceptible de causer des problèmes psychologiques importants. Il est donc recommandé de demander de l'aide.
Le syndrome MRKH a une incidence mondiale de 1 naissance sur 4 500 de femmes, selon diverses études.
Il est généralement diagnostiqué plus fréquemment à l'adolescence, quand il est vérifié si le cycle menstruel se développe. Bien que les femmes ne puissent pas avoir d'enfants, leurs ovaires sont normaux et fonctionnels.
Symptômes du syndrome de Rokitnasky-Küster-Hauser
Les symptômes du syndrome MRKH varient considérablement d'une femme à l'autre, gardez donc à l'esprit que les femmes affectées peuvent ne pas présenter tous les symptômes mentionnés ci-dessous.
Syndrome de Mayer-Rokitansky-Hauser de type I
Ce type est également connu sous le nom d'aplasie de Müller et se caractérise par un développement inadéquat de l'utérus et du vagin. Dans la plupart des cas, l'utérus et / ou le vagin ne se sont pas développés (aplasie); ou il y a un rétrécissement de la partie supérieure du vagin et de l'utérus (atrésie). Les trompes de Fallope peuvent également être affectées.
Certaines des caractéristiques ou symptômes du syndrome MRKH sont:
-Première ménopause ou absence de règles pendant la puberté. Bien que ce symptôme soit courant dans le syndrome MRKH, le patient subit une puberté avec un telarca normal et une adrénarche. Cependant, il n'y a pas de menstruation.
La fonction des ovaires étant normale, les patients subissent tous les changements associés à la menstruation ou à la puberté.
Organes génitaux externes normaux.
- profondeur vaginale réduite, de 2 à 7 cm.
- Caractéristiques sexuelles telles que les seins normaux et les poils pubiens.
-Ovaires fonctionnels avec des niveaux normaux d'oestrogène.
-Modèles chromosomiques normaux.
-Dans le syndrome MRKH de type 1, seuls le vagin et l'utérus sont anormaux. Dans le type II MRKH, des défauts dans le vagin et l'utérus peuvent également être accompagnés par des anomalies dans le tube Fallope, comme dans les reins ou la colonne vertébrale.
-Bien que les anomalies cardiaques soient rares, la fenêtre aorto-pulmonaire, le défaut septal auriculaire et la sténose de la valve pulmonaire peuvent survenir
-Les patients atteints du syndrome MRKH se plaignent généralement de douleurs abdominales cycliques dues au détachement de l'endomètre cyclique sans voie de drainage brevetée.
-Estilité: De nombreux patients recherchent souvent des soins cliniques pour l'infertilité, mais pas pour l'aménorrhée primaire.
- Un autre symptôme est la difficulté des relations sexuelles, car le degré d’aplasie vaginale peut varier d’une absence complète à un développement médiocre, ce qui peut entraîner une dyspareunie.
-Difficultés de miction, incontinence urinaire, infections urinaires récidivantes.
- anomalies vertébrales.
-Une écharpe tissulaire palpable peut être présente au niveau de la réflexion péritonéale.
- malformations rénales.
Les symptômes du syndrome MRKH de type II
L'insuffisance rénale est l'anomalie la plus fréquente associée au syndrome MRKH de type II.
Les femmes avec le type de syndrome MRKH II peuvent ne pas avoir les reins, malformation d'un ou les deux reins (dysplasie rénale), le développement des reins (hypoplasie) et / ou placement incorrect dans le corps de l'un ou les deux reins (de ectopie rénale).
Ces anomalies peuvent causer une carence en croissance rénale, des calculs rénaux, une sensibilité accrue aux infections des voies urinaires et de l'accumulation anormale de l'urine dans les reins en raison de l'obstruction.
Beaucoup de femmes atteintes du syndrome MRKH de type II peuvent aussi avoir des malformations squelettiques telles que des problèmes osseux dans les vertèbres cervicales et thoraciques qui peuvent se développer de manière incorrecte.
Les anomalies de la face peuvent également se produire, par exemple ayant une mâchoire anormalement faible (de micrognathia), bec de lièvre, et le sous-développement d'un côté de la face, ce qui entraîne une asymétrie faciale.
De nombreuses femmes atteintes peuvent également développer des problèmes d'audition, principalement dus à des anomalies structurelles de l'oreille moyenne.
Lorsque les oreilles sont impliquées, le trouble peut être appelé syndrome de l'oreille rénale génitale (GRES).
Certaines des femmes atteintes du syndrome de MRKH de type II ont présenté des anomalies physiques supplémentaires, notamment des défauts dans les mains et / ou les bras.
Ces anomalies des membres peuvent inclure l'absence d'un ou plusieurs doigts et les orteils, le pouce en double et l'absence d'os de l'avant-bras long et mince (rayon absent). Tous les symptômes ne surviennent pas chez tous les patients, cela dépend de l'emplacement et de la gravité.
Les causes
Dans la plupart des cas, l'origine du syndrome MRKH est inconnue et survient chez les femmes sans antécédents familiaux.
Les chercheurs soulignent que des facteurs génétiques et environnementaux sont à l’origine du syndrome, même si aucun gène ni aucun gène associé à cette condition n’a encore été déterminé.
Historiquement, les chercheurs ont suggéré que le syndrome peut survenir à la suite d'une maladie ou d'une exposition fœtale maternelle à diverses substances nocives, telles que certains médicaments, l'abus de drogues, l'alcool ...
Cependant, aucune étude ne corrobore cette association entre le syndrome et la consommation d'autres médicaments.
Dans certaines familles, le syndrome semble avoir un mode de transmission dominant. Cependant, le modèle d’héritage est difficile à établir, car toutes les femmes qui en souffrent n’ont pas les mêmes symptômes, même si elles appartiennent à la même famille.
Selon les chercheurs, il s'agit très probablement d'une combinaison de facteurs génétiques et environnementaux.
Quant aux anomalies du processus de reproduction, elles sont dues à un développement incomplet du canal de Müller, mais sa cause reste inconnue.
Il semble que ces dernières années, il y a eu une augmentation des preuves, en ce sens que le syndrome de MKRH est une maladie génétique. L'augmentation des études de cas a permis de renforcer cette idée.
Nous parlons de troubles génétiques lorsqu'il existe une combinaison de gènes pour un trait particulier. Dans le cas où il s'agirait d'une maladie génétique dominante telle qu'elle serait dans ce cas, elle serait produite par le développement anormal d'une copie unique d'un gène anormal.
Ce gène défectueux peut être hérité à la fois par la mère et par le père ou par une nouvelle mutation du gène lui-même.
La probabilité de transmettre ce gène défectueux sera donc de 50% pour chaque grossesse, quel que soit le sexe de l'enfant.
L'héritage polygénique a également été proposé comme cause du syndrome MRKH.
À ce jour, sept délétions et une duplication de segments chromosomiques ont été trouvées chez plusieurs personnes atteintes du syndrome MRKH. Mais une seule de ces anomalies a été trouvée par personne.
Actuellement, ces chromosomes ont été identifiés là où des éliminations segmentaires sont possibles. Ces chromosomes sont: chromosome 1 (1q21.1), 4 (4q34q), 8 (8p23, 1), 10 (10p14-15), 16 (16p11.2), 17 (17q12) et 22 (22q11.21) , et la duplication a été trouvée sur le chromosome X (Xpter-p22.32).
Toutes ces nouvelles informations a conduit les chercheurs à sélectionner plusieurs gènes candidats, y compris: HNF1B, LHX1, TBX6, ITIH5 et SHOX.
Traitement
L'objectif du traitement est que le patient ait un fonctionnement sexuel complet et satisfaisant.
Les symptômes étant très variables, la collaboration d'une équipe de spécialistes est nécessaire pour garantir une approche globale du traitement.
En général, ils sont encouragés les patients atteints du syndrome MRKH à chercher de l'aide psychologique après le diagnostic et avant le traitement, étant donné que ce syndrome peut causer de l'anxiété ou la détresse psychologique. Des groupes de soutien sont également recommandés.
En ce qui concerne le traitement de l'aplasie vaginale, il faut créer un nouveau vagin.
Le traitement peut être chirurgical ou non. Le non-chirurgical sera toujours la première option. Une des options sont les dilatateurs vaginaux, qui aident à agrandir ou à créer un vagin.
La méthode la plus connue est le dilatateur Franck, également connu sous le nom de dilatateur périnéal, qui ne nécessite aucune intervention chirurgicale.
Être auto-administré le patient doit être suffisamment motivé pour l'utiliser. Il dure environ six semaines à plusieurs mois.
Une autre option peut être une vaginoplastie, qui créerait un vagin peu profond. Mais à propos de cette chirurgie, il n'y a pas beaucoup d'accord sur les techniques à utiliser.
La technique Mclndoe est l'une des interventions chirurgicales les plus utilisées pour la reconstruction du vagin. Une greffe de peau prélevée sur les fesses ou la cuisse est prélevée et appliquée sur une prothèse en forme de pénis qui est gonflable. Cette prothèse de greffe avec les moules à tunnel vaginal, ce qui ouvre avec un scalpel et disséquée en coupe et de façon franche, avec soin de ne pas endommager le vegija, du rectum ou du péritoine.
Il est laissé sept à dix jours et quand il est changé, il est sous anesthésie. Plus tard, le patient utilisera d'autres prothèses de dilatation. À trois mois, le patient peut avoir des relations sexuelles.
Le problème avec cette technique est qu’en plus de laisser des cicatrices, une dilatation à long terme est nécessaire.
Une autre technique utilisée pour le syndrome MRKH est le néo-vagin intestinal.
Cette technique utilise un segment isolé des intestins. Il consiste à sectionner un fragment du côlon sigmoïde par une incision abdominale et à le transférer dans la zone où l’espace du néo-vagin a été créé. Cette technique est assez complexe et peut durer jusqu'à 8 heures.
L'avantage de cette technique est qu'elle laisse un long vagin de longueur adéquate et qu'elle est autolubrifiante. En ce qui concerne les inconvénients, la première chose serait qu'il s'agisse d'une chirurgie complexe et infrabominale, qui laisse des sécrétions muqueuses excessives à travers le fragment intestinal, un prolapsus vaginal muqueux et une inflammation de la muqueuse dans la néovagine.
Enfin, une autre des techniques est la Technique Vecchieti.
Cette technique exerce une pression progressive continue par une olive acrylique à travers la puissance de l'espace néovaginal et la paroi abdominale. Ensuite, un dispositif de traction est placé dans la cavité péritonéale et élimine progressivement la voûte vaginale. Cette technique est maintenant réalisée par laparoscopie.
Références
- https://visualsonline.cancer.gov/
- https://ghr.nlm.nih.gov/condition/mayer-rokitansky-kuster-hauser-syndrome
- http://emedicine.medscape.com/article/953492-overview
- http://www.news-medical.net/health/Mayer-Rokitansky-Kuster-Hauser-(MRKH)-Syndrome-Symptoms-(Spanish).aspx
- http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=3109
- https://rarediseases.org/rare-diseases/mayer-rokitansky-kuster-hauser-syndrome/
- http://www.biomedcentral.com/content/pdf/1750-1172-2-13.pdf
- Image source: http://www.mrkhnorge.org/mrkh/?lang=fr