Partager:



Syndrome de Pallister-Killiam Symptômes, causes, traitement
Le Syndrome de Pallister-Killian (SPK), également connu sous le nom de tétrasomie 12, est une maladie rare d'origine génétique qui se caractérise par un large spectre d'atteinte de plusieurs organes.
Cliniquement, cette condition est définie par un retard mental, un retard psychomoteur, hypotonie, phénotype facial atypique, anomalies pigmentaires de la peau et l'alopécie (Toledo-Bravo Laguna, Champ-Casanelles, Santana-RODRIGUEZ, Santana -Artiles, Sebastían-Garcñua, Cabrera-López, 2014).
En outre, d'autres types de complications médicales peuvent également apparaître liées à des malformations de différents systèmes corporels ou d'épisodes convulsifs (Toledo-Bravo de la Laguna et al., 2014).
L'origine étiologique de cette maladie est associée à une altération génétique répartie en mosaïque. Plus précisément, il est dû à la présence d'un chromosome 12 supplémentaire dans certaines cellules de l'organisme (Understanding Chromosome Disorders, 2016).
Le diagnostic du syndrome de Pallister-Killiam peut être posé à la fois au stade prénatal et postnatal. L'objectif principal est l'identification des caractéristiques cliniques et l'utilisation d'une étude génétique de confirmation (Méndez, Rodríguez, Boluarte, Cartolin, Valdéz et Matheus, 2013).
Ce syndrome a un taux de mortalité élevé (Ramírez Ferández, García Cavazos, Sánchez Martínez, 2007). Cependant, l'approche pharmacologique médicale et le traitement de réadaptation peuvent apporter des avantages importants dans la qualité de vie et l'état clinique des personnes touchées (Méndez et al., 2013).
Caractéristiques du syndrome de Pallister-Killiam
Le syndrome de Pallister-Killiam (SPK) est un type de maladie génétique de la mosaïque. Dans ce cas, l'altération chromosomique n'affecte que certaines cellules de l'organisme.
Certaines institutions, telles que Genectis Home Reference (2016), classent cette pathologie dans les troubles du développement.
À un niveau général, les troubles du développement ou les troubles du développement à l’échelle internationale se réfèrent généralement à un large éventail d’altérations et d’anomalies physiques et cognitives. Tous ces facteurs entraînent une déviation ou un retard important du développement par rapport aux profils normaux ou attendus (Institut national des troubles neurologiques et des accidents vasculaires cérébraux, 2015).
Dans le syndrome de Pallister-Killiam, une large affectation de différents systèmes et organismes est identifiée (Genectis Home Reference, 2016).
Il se caractérise principalement par un retard mental, hypotonie musculaire, développement des traits du visage distinctifs, la pigmentation anormale de la peau ou la croissance des cheveux, entre autres anomalies congénitales (Genectis Accueil de référence, 2016).
De plus, le syndrome de Pallister-Kiliam est une origine de la maladie congénitale rare (Turleau, 2009) qui peut recevoir un grand nombre de noms dans la littérature médicale (Organisation nationale pour les maladies rares, 2016):
- Syndrome de mosaïque Pallister-Killiam.
- Syndrome de l'ischromosome 12p.
- Syndrome de Killiam.
- Syndrome de Nicola-Teschler
- Syndrome de mosaïque pallistif.
- Tetrasomy 12p.
- Syndrome de Killiam-Tescheler-Nicola.
Cette maladie a été initialement décrite par Pallister en 1977 (Toledo-Bravo de la Laguna et al., 2014).
Dans les premières publications, il a noté deux cas de patients adultes dont le cours a été marquée par plusieurs constats: convulsions, hypotonie musculaire, déficit intellectuel, des malformations musculo-squelettiques et configuration organique, du visage grossier et les changements de couleur de la peau (et Méndez al., 2013).
Parallèlement, Teschler-Nicola et Killiam en 1981 ont décrit le même tableau clinique chez une fillette de trois ans (Méndez et al., 2013).
Par conséquent, dans les premiers rapports cliniques de référence, il est fait largement à une condition médicale caractérisée par la combinaison des crises, un retard mental et un phénotype caractéristique physique (Toledo-Bravo Laguna et al., 2014).
De plus, et en 1985, il a réussi à identifier premier cas Gilgenkratz pendant la grossesse, quelque chose aujourd'hui commun grâce à des techniques modernes de diagnostic (Méndez et al., 2013).
Statistiques
Les chiffres de prévalence du syndrome de Pallister-Killiam ne sont pas connus avec précision. Peu de diagnostics définitifs ont été réalisés et la plupart d'entre eux n'ont pas été publiés dans la littérature médicale (Understanding Chromosome Disorders, 2016).
Ainsi, tous les auteurs et institutions définissent ce syndrome comme une pathologie génétique rare ou rare dans la population générale (EuRed, 2016).
Il y a environ 15 ans, le syndrome Pallister-Killiam avait été identifié dans une centaine de cas seulement dans le monde. Actuellement, ce chiffre a dépassé 200 personnes touchées (Understanding Chromosome Disorders, 2016).
Les enquêtes épidémiologiques ont estimé l’incidence de cette maladie chez environ 5,1 cas par million de nouveau-nés (Understanding Chromosome Disorders, 2016), bien que des auteurs tels que Toledo-Bravo de la Laguna et ses collaborateurs (2014) la placent à 1 / 25 000.
Une prévalence plus élevée associée aux caractéristiques sociodémographiques des personnes touchées n'a pas été identifiée. Le syndrome Pallister-Killian peut apparaître dans n'importe quel sexe ou groupe technique et / ou racial.
Signes et symptômes
Dans l'évolution clinique du syndrome de Pallister-Killian, une grande variété de signes et de symptômes peuvent être identifiés. Tous associés à des anomalies craniofaciales et / ou à des altérations du muscle squelettique et cognitives.
Paramètres du visage
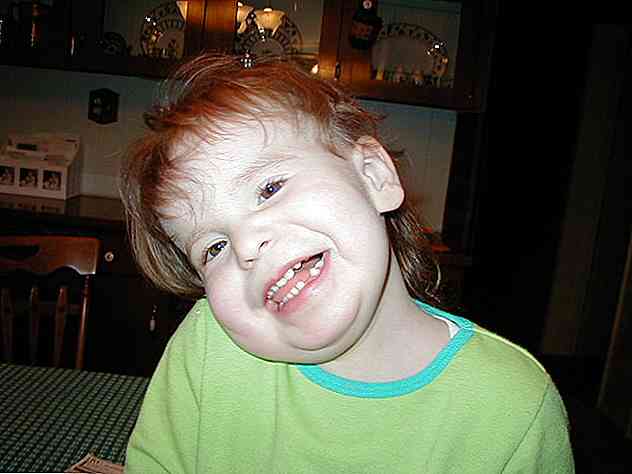
Le développement de malformations cranio-faciales de la phase de gestation à la croissance postnatale et infantile constitue l'un des signes médicaux les plus caractéristiques du syndrome de Pallister-Killiam.
Les signes et symptômes les plus courants comprennent des anomalies dans les différentes structures crâniennes et faciales qui donneront lieu à une apparence rugueuse et atypique (Toledo-Bravo de la Laguna et al., 2014; Understanding Chromosome Disorders, 2016):
- Brachycéphalie: ce terme se réfère à une configuration crânienne qui entraîne une augmentation de la largeur de la tête et un aplatissement des zones occipitales et postérieures.
- Configuration crânienne frontale: Les zones antérieure et frontale de la tête ont tendance à se développer plus que d'habitude. Un front proéminent ou bombé peut être vu.
- Configuration crânienne postérieure: la zone la plus postérieure de la tête semble présenter un état sous-développé. Un occiput plat peut être vu.
- Hypertherisme: les yeux doivent être placés plus loin que d'habitude. Sur le plan visuel, les yeux sont largement séparés.
- Configuration nasale: Le nez a généralement un grand volume, avec un pont racine ou large. Les narines doivent être orientées vers l'avant (narines antéversées).
- Configuration orale et maxillaire: les structures orales doivent présenter une taille anormale. La mâchoire est plus petite que d'habitude (micrognathie). La lèvre supérieure prend un aspect mince et réduit, tandis que la lèvre inférieure est épaisse. La langue est plus grande que prévu et le long sillon nasolabial.
- Pavillons Auditifs: les oreilles ont une position basse et sont tournées vers l'arrière.
- Alopécie:La croissance des cheveux est anormale dans différents domaines. Le plus commun consiste à observer de petites zones de calvitie dans les sourcils, les cils ou la tête.
- Taches acromiques et hyperchromiques: Il est possible d'identifier le développement de petites taches sur les zones du visage. Ils se caractérisent par une perte de couleur ou d'apparence sombre.
Malformations musculo-squelettiques
Bien qu’elles soient moins importantes que les altérations faciales, il est très fréquent d’observer plusieurs anomalies musculo-squelettiques chez les patients atteints du syndrome de Pallister (Comprendre les troubles chromosomiques, 2016):
- Cou: La distance entre la tête et le tronc du corps est généralement réduite. Au niveau visuel, nous pouvons voir un cou court ou plus petit que d'habitude.
- Colonne vertébrale: Bien qu'il ne soit pas très courant d'identifier les modifications de la colonne vertébrale, il est possible que le spina bifida, l'appendice sacré, la scoliose ou la cyphose apparaissent.
- Astuces: Les bras et les jambes présentent également une croissance anormale, inférieure à celle attendue pour le sexe et l’âge biologique de la personne atteinte.
- Polydactylie: des altérations liées au nombre de doigts et d'orteils peuvent également apparaître. Le plus commun est de voir plus de doigts sur les mains
Hypotonie musculaire et retard psychomoteur
Les anomalies liées à la structure musculaire et à la mobilité sont une autre des caractéristiques cliniques cardinales du syndrome Pallister-Killian (Comprendre les troubles chromosomiques, 2016):
Hypotonie musculaire se réfère à l'identification d'un tonus musculaire anormalement réduit ou de tension. Au niveau visuel, la flaccidité et la labilité peuvent être observées dans différents groupes musculaires, en particulier dans les extrémités.
Ainsi, la pathologie musculaire et squelettique entraînera un retard important dans l'acquisition de différentes habiletés motrices, à la fois pendant la période néonatale et pendant l'enfance.
Bien que les périodes de développement soient variables parmi celles touchées, le calendrier le plus courant comprend les étapes suivantes:
- Sedestación: la capacité à acquérir des postures indépendamment, à s'asseoir ou à tourner avec votre propre corps peut commencer à se développer à partir de 3 mois. Cependant, chez les personnes touchées par ce syndrome, il peut être retardé jusqu'à l'âge de 8 ans.
- Premiers pas: l’habitude est que les enfants commencent à faire leurs premiers pas vers 12 mois, cependant, dans cette pathologie, cette étape de l’évolution peut être retardée jusqu’à 9 ans. De plus, dans de nombreux cas, certaines méthodes compensatoires telles que les attelles ou les chaussures spécialisées sont essentielles.
Altérations neurologiques
Le système nerveux est une autre des zones fortement touchées. Dans la plupart des cas, les signes et les symptômes sont principalement liés à des convulsions et à une déficience intellectuelle (Toledo-Bravo de la Laguna et al., 2014; Understanding Chromosome Disorders, 2016):
- Saisies: la présence et le développement d'une activité électrique neuronale inhabituelle, altérée et désorganisée peuvent entraîner la présence d'événements récurrents définis par des spasmes musculaires, une agitation motrice ou une absence de conscience. La structure cérébrale est gravement altérée, entraînant une détérioration cognitive et tissulaire importante.
- Déficience intellectuelle: Bien que le niveau de déficience cognitive soit variable, dans la plupart des cas, un quotient intellectuel faible ou limite est identifié. Les zones les plus touchées sont le psychomoteur et le linguistique, les critères cliniques du trouble du spectre autistique remplissant l'une des zones touchées.
- Retard généralisé dans le développement: le rythme d'apprentissage des différentes compétences quotidiennes et académiques est généralement lent dans une bonne partie des personnes touchées. Des adaptations et un soutien scolaire spécialisé sont généralement requis.
Autres anomalies
Bien qu'ils soient moins fréquents, d'autres types de complications médicales peuvent également apparaître (Organisation nationale des maladies rares, 2016) (Toledo-Bravo de la Laguna et al., 2014):
- Anomalies et malformations cardiaques, gastro-intestinales, rénales et génitales.
- Sténose auditive
- Hypoplasie pulmonaire.
- Strabisme et cataracte.
- Réduction de l'acuité visuelle et auditive.
Les causes
L'origine du syndrome de Pallister-Killian est associée à une anomalie génétique de la mosaïque sur le chromosome 12. Elle n'affecte que le matériel génétique de certaines cellules de l'organisme (Inage et al., 2010).
Les chromosomes font partie du noyau de toutes les cellules présentes dans le corps humain. Ils sont composés d'une grande variété de composants biochimiques et contiennent les informations génétiques de chaque individu (National Organisation for Rare Disorders, 2016).
Les humains ont 46 chromosomes différents, organisés par paires et numérotés de 1 à 23. De plus, au niveau individuel, chaque chromosome a une aire ou un bras court appelé "p" et un autre long appelé "q" (Organisation nationale des maladies rares). 2016).
L'anomalie affecte le chromosome 12 et entraîne la présence d'un chromosome à structure anormale, appelé isocromosome (Genetics Home Reference, 2016).
Ainsi, ce chromosome a tendance à avoir deux bras courts au lieu d'un de chaque configuration p (court) et long (q) (Genetics Home Reference, 2016).
En conséquence, la présence de matériel génétique supplémentaire et / ou anormal modifiera le cours normal et efficace du développement physique et cognitif de la personne atteinte, en donnant lieu aux caractéristiques cliniques du syndrome Pallister-Killian (Genetics Home Reference, 2016). .
Diagnostic
Le syndrome pallisto-killien peut être identifié pendant la grossesse ou au stade postnatal, en fonction des caractéristiques cliniques et des résultats des différents tests de laboratoire (Turleau, 2009).
Pendant la grossesse, les tests les plus fréquemment utilisés sont l'échographie, l'amniocentèse ou le prélèvement de villosités choriales (Turleau, 2009).
En ce sens, l'analyse du matériel génétique de l'embryon peut nous fournir une confirmation de cette pathologie, à travers l'identification d'anomalies compatibles (Turleau, 2009).
En revanche, si le diagnostic est posé après la naissance, il est fondamental (Understanding Chromosome Disorders, 2016):
- Biopsie cutanée.
- Analyse de sang
- Etude des lymphocytes sanguins.
- Hybridation fluorescente in situ.
- Hybridation génomique comparative.
Traitement
Aucune thérapie spécifique n'a été conçue pour le traitement des personnes atteintes du syndrome de Pallister-Killian (Organisation nationale des troubles rares, 2016).
Le syndrome de Pallister-Killian est généralement associé à un mauvais pronostic et à des taux de mortalité élevés (Ramírez Ferández, García Cavazos, Sánchez Martínez, 2007).
Cependant, le traitement de réadaptation, l’éducation spéciale et l’ergothérapie peuvent offrir un bon pronostic fonctionnel et améliorer la qualité de vie des personnes touchées.
Par exemple, Méndez et son équipe (2013) décrivent un cas de traitement de réadaptation caractérisé par:
- Amélioration des capacités psychomotrices: contrôle de la tête, assise et position indépendantes.
- Amélioration du niveau de vigilance, de l'attention, de la régulation comportementale.
- Amélioration de la motricité fine, telle que la pression manuelle.
- Emission de sons et sourire contextuel.
- Suivi visuel, fixation et discrimination des stimuli auditifs.
Références
- Ecured. (2016). Syndrome de Pallister-Killian. Récupéré d'Ecured.
- Genetics Home Reference. (2016). Syndrome de mosaïque pallistero-killienne. Récupéré de Genetics Home Reference.
- Inage et al. (2010). Chevauchement phénotypique de la trisomie 12p et du syndrome de PallistereKillian. European Journal of Medical Genetics, 159-161.
- Méndez, M., Rodríguez, M., Boularte, A., Cartolin, R., Valdéz, G. et Matheus, F.(2013). Syndrome de Killian-Pallister. Rapport d'un cas en thérapie de rééducation interdisciplinaire. Rev Med Hered.
- NORD. (2016.). Syndrome de Pallister Killian Mosaic. Récupéré de l'Organisation nationale des maladies rares.
- Ramírez-Fernández, M., García Cavazos, R. et Sánchez-Martínez, H. (2007). Syndrome de Pallister-Killiam. Communication d'un cas. Ginecol et obsta Mex, 414-18.
- Toledo-Bravo de la Laguna, L., Campo-Casanelles, M., Santana-Rodriguez, A., Santana-Artiles, A., Sebastian-Garcia, I., et Cabrera-Lopez, J. (2014). Présentation de trois cas de syndrome de Pallister-Killiam. Rev Neurol, 63-68.
- Turleau, C. (2009). Tetrasomy 12p. Obtenu chez Orphanet.
- Comprendre les troubles chromosomiques. (2016). Syndrome Pallister-Killiam. Comprendre les troubles chromosomiques.
- Image source